Accelerating Innovative Cell Therapy Development Through Phase-Appropriate Potency Control



Cell therapies have emerged as a transformative tool of modern medicine, offering unprecedented potential to treat and cure a wide range of diseases. Engineered cell and gene therapies are able to address the etiologic genetic mutation or eradication of the disease-relevant cellular compartment, with profound improvements in clinical outcomes. From immune-based approaches like T cell therapy for cancer to regenerative applications utilizing stem cells, cell therapies are redefining the boundaries of treatment modalities.
Gene delivery technologies enable the introduction, deletion, or modification of genetic material within cells, equipping them with novel therapeutic properties or optimizing their natural capabilities. From viral vectors such as lentiviruses and adenoviruses to non-viral methods like electroporation and lipid nanoparticles, these technologies form the backbone of genetic engineering in cell-based treatments.
The quality control measures that underpin the development and commercialization of these promising therapeutics are key to furthering them within the larger biopharmaceutical pipeline. In particular, potency assays are central to this pursuit, as these analytics are crucial to ensuring product consistency, efficacy, and safety. The potency of a product is the specific ability or capacity of a product to achieve a defined biological effect. Potency assays are quantitative measures of biological activity and are typically assessed in vitro.
In the case of CAR or TCR T cell products, both the vector to deliver the gene of interest (GOI) and the gene-modified T cell drug product require potency assays to be in place to support product release and stability. By adopting a phase-appropriate yet prospectively considered approach to potency development as early as possible in a process, organizations can arrive at a potency control strategy that improves the foundational understanding of a product’s quality and consistency and results in a strategy that will be suitable for a marketing application while not jeopardizing the use of valuable clinical data needed to support the safety and efficacy assessment for the application.
Genetic medicine potency assays
The development of potency assays can be challenging due to the complex nature of cell and gene therapies and the lack of standardized methods in the broader development space. Development of suitable and robust potency methods requires plenty of development data and correlation from orthogonal readouts. During early phases of drug development, a potency assay can be a quick and simple method suitable for the phase. However, through the course of drug development, potency assays often require several rounds of iteration and maturation, including implementation of controls and standards. Moreover, the functional potency assays that support a marketing application’s overarching potency strategy must be able to effectively measure a product’s mechanism of action (MOA) or biological function. For many complex products, the understanding of the drug MOA evolves through the course of development. It is therefore recommended that the potency work should start early during development.
The assay development can come at a significant cost as the assays may require several custom reagents, including the need for establishing cell banks and reference materials. Developing a potency strategy for genetic medicines is often challenging for the companies pioneering these treatments, many of whom are working with small teams, constrained resources, and competing priorities throughout development.
Regulatory expectations for potency assays
The existing successes of CAR and TCR T cell products mean that the regulatory expectations around these products are reasonably well documented1,2,3 — including that potency assays should:
- Reflect biological effects that represent the proposed clinical MOA
- Characterize a product well enough to identify and evaluate the impact of process changes
- Enable operators to establish criteria for stability and comparability during process changes, improvements, and lot release.
Pre-Clinical Development to FIH
- Establish proof of concept
- Initiate development of multiple readouts: genetic and protein
- Semi-quantitative with phase-appropriate specificity and sensitivity
- Evaluate suitability for in vitro and animal model testing

Later Phases of Clinical Development To Pivotal
- Refine assays for quantitative readouts based on early clinical data: Identify Reference standards and critical reagents
- Develop MoA functional potency
- Qualify assays for accuracy, precision, and robustness
- Assess suitability for later phases: Establish acceptance criteria

Toward Commercial Filing
- Further optimize assay based on expanded clinical data
- Validate with larger sample size and routine handling conditions
- Finalize documentation for regulatory submission
- Confirm method acceptance criteria
Figure 1: An overview of the key considerations for a phase-appropriate potency strategy.
The regulatory agencies suggest potency assays be in place even during the initial phases of development so that, by the time the product has moved into pivotal efficacy studies, quantitative potency assays that measure MOA-reflective biological activity are required for lot release and stability. The latest FDA guidance1 emphasizes a lifecycle approach to potency that is grounded in quality risk management, where potency tests are considered throughout the product lifecycle, from product development all the way to product licensure, and can adapt with gained knowledge of mechanism of action and assay experience.
Although these requirements are widely acknowledged, many companies run into snags early when it comes to approaching potency. For example, an organization can focus exclusively on potency for the drug product without recognizing that the vector is also considered a critical component that furnishes a pharmacological activity to the drug product and should include testing of biological activity. Or an organization may not have the bioassay development expertise or the regulatory experience to develop the potency control strategies in a phase-appropriate manner. Engaging with a full-service CDMO with sophisticated analytical capabilities, expertise, and infrastructure can help expedite the development of potency control strategies.
Furthermore, while early development potency lot release assays can be less stringent “litmus tests,” these analytics are likely to eventually need to have two-sided acceptance criteria. Developers must also consider the type of statistical analysis they perform, such as parallel line analysis for more complex assays at later phases to demonstrate similarity to a reference material. Establishing and maintaining a reference material is ideal as these provide a consistent point of reference to compare the biological activity of a drug product or substance, ensuring accurate and reliable relative potency measurements throughout development.
Companies that deprioritize development of methods to measure biological potency until later phases of development risk falling behind in maturing their assays effectively and can encounter regulatory and technical setbacks as a program progresses. This can make it hard for organizations to pinpoint challenges, even for those that have retained earlier samples for testing, as the quality and stability of these retains cannot be assured.
Creating a balanced potency assurance strategy
CAR and TCR T cell therapies affect target cells in an antigen-specific manner using multiple mechanisms, and therefore the use of orthogonal methods is recommended. A CAR or TCR T cell product is made by delivering the GOI using a suitable vector, e.g., a lentiviral vector (LVV). Upon GOI delivery, the engineered receptor is expressed on the T cells that can bind to specific antigens on target cells (e.g., cancerous cells). When the therapeutic cells interact with target cells via the engineered CAR or TCR, intracellular signaling cascades within the DP cells leads to the release of pro-inflammatory cytokines, cytolysis of the target cells, and expansion and proliferation of the engineered cells. These are key indicators of T cell activation. Thus, interferon gamma (IFNγ), a pro-inflammatory cytokine, serves as a critical downstream marker in this cascade, making it a relevant attribute for the MOA and quantifiable readout of CAR function4.
When measuring potency for these complex therapeutics, a set of potency assays has been well validated for these applications (Figure 2). They include:
- Measuring the delivery and integration of GOI at genetic level: This can be done by using molecular techniques like ddPCR or qPCR.
- Measuring the expression of transduced GOI at a protein level: Transgene expression can be measured using flow cytometry to quantify the percentage of cells expressing the CAR.
- The biological activity of the GOI can be measured by quantifying cytokine release using cell-based assays such as ELISA, ELLA, MSD, or flow cytometry.
- The biological activity of the GOI can also be measured by quantifying the killing of target cells using cell-based assays leveraging luminescence or flow cytometry.
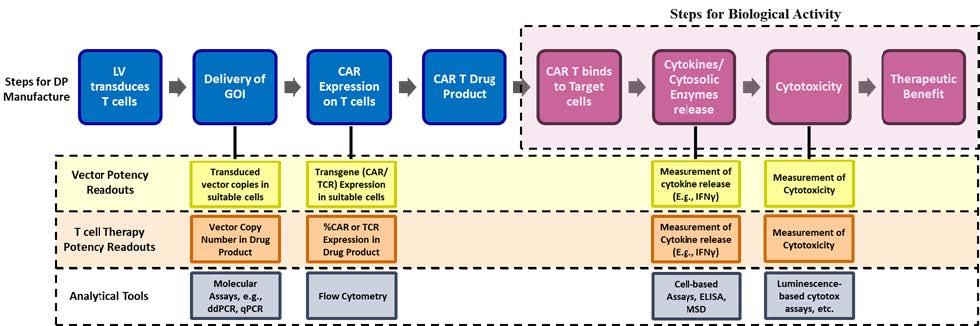
Figure 2: A simplified overview of the gene-edited cell therapy manufacturing process and potency assay strategy based on key process steps.
In the example of a CAR or TCR T cell therapy, expression assays for the GOI are necessary for understanding potency for early-stage processes. Expression assays alone do not offer insight into the biological function of the cell product, however. This is where assays like those used for cytokine release or cytotoxicity are integral to a program and why at least one should be incorporated early, even if they are not used as qualified release assays at this nascent stage of development. These tests require more up-front work for method development — establishing acceptance criteria and creating controls and reference standards — but they can offer greater insight that is indispensable in the long term.
Ideally, biological potency assays can be introduced early in development to gain product and assay knowledge critical to enabling the most appropriate methods and acceptance criteria for release and stability testing during pivotal clinical trials. Additionally, these assays are extremely useful to have in analytical comparability studies, including for changes introduced in early development.
The vector potency determination also takes a similar approach of analytical readouts (Figure 2). During the initial phases of development for these therapies, the primary focus is on verifying the ability of the vector to successfully deliver the GOI into representative cells. This approach typically employs transduction of a target cell line by the vector. The transduced cells are cultured, harvested, followed by PCR amplification of the integrated provirus sequence, offering a precise measurement of the delivered gene copy. The functional vector potency readout is designed to demonstrate the ability of the vector to generate a biologically active CAR or TCR T cell based on readouts like cytokine release.
Key considerations during development of potency methods
Method development and optimization for these therapies require a systematic approach to ensure robust and reproducible processes that deliver high-quality therapeutic products. The molecular methods measuring drug product potency are based on accurate and precise quantification of cells with integrated vector based on a PCR-based readout. The development and qualification of a molecular assay are relatively straightforward and focus mainly on optimizing the primers, probe and PCR conditions, plus ensuring appropriate method controls. Similarly, the measurement of %CAR or %TCR -positive cells in DP using flow cytometry requires identification of appropriate antibodies and optimizing the staining conditions and gating strategy.
The biological potency for T cell product can be based on in vitro cell-based methods aimed at measuring cytokine release and/ or cell cytotoxicity. The method setup requires activation of T cell drug product by co-culture with antigen-presenting target cells or incubating with target antigen. Establishing the critical reagents, most importantly the target cell line or target antigen, is the first step to developing a robust method. Several pros and cons must be considered when finalizing the choice for the activation step as it must demonstrate consistent response in the potency readout. The target cell lines must be comprehensively tested to confirm cell viability, genetic stability, and expression of the specific antigen at appropriate levels.
Optimization of the method variables such as cell seeding density, effector-to-target cell ratio during co-culture, duration of culture, etc., are equally critical to building a robust method. Part of the puzzle also requires identifying and optimizing a suitable analytical readout. For a cytokine release potency, the typical analytical tools include ELISA, ELLA, MSD, etc. The qualities influencing the choice of readout often include accuracy, precision, and robustness of the method, operator hands-on time, degree of automation, availability, and cost of suitable kits and reagents.
The analytical tools and readouts used for vector potency measurement are similar to the T cell therapy product, but the key difference is the design, setup, and reportable results determining potency. A vector potency setup typically uses representative non-transduced cells, either derived from healthy donors or a suitable cell line, that are transduced in small-scale format by titrating vector test article. Upon completion of culture, the cells are harvested and tested to measure the transduction ability of the vector, plus downstream function, such as cytokine release, of the delivered GOI.
Generating and characterizing cell banks is critical as it serves as a consistent and reliable starting material for transduction. Equally critical is bridging and demonstrating comparability of cell banks when needed for ensuring method consistency through the product’s lifecycle. To assess vector dose response, dose titration studies are conducted to correlate vector concentration with functional outcomes, such as transgene expression or biological activity4. Variables such as multiplicity of infection (MOI) range, transduction conditions such as exposure time, and media composition, are carefully evaluated to optimize gene transfer and maintain cell health. Post-transduction, harvest conditions, including timing and cell viability, must be standardized for a robust and consistent method performance.
Summary
Overall, early development of robust potency strategies is crucial for ensuring the clinical and regulatory success of advanced therapies. By integrating phase-appropriate assays and continuously refining methods based on evolving understanding of mechanisms of action, developers can mitigate risks and improve product consistency. The establishment of reliable reference materials and the use of orthogonal testing approaches likewise provide critical insights into a product’s biological activity, facilitating smoother regulatory submissions. Ultimately, a well-defined potency strategy supports the timely delivery of quality, safe, and effective therapies while minimizing setbacks and aligning with regulatory expectations.
Many sponsors face challenges developing a potency control strategy specifically for cell and gene therapies due to the complexities in understanding and measuring the biological effect of the products. Additionally, smaller, younger companies may not have the required resources and expertise, or a larger organization may be working on more traditional modalities and not have the CGT experience. With extensive experience across a range of modalities—including genetically modified cell therapies, gene editing, and viral and non-viral vectors—ElevateBio ensures that potency assays evolve appropriately throughout the development lifecycle, from preclinical stages to first-in-human trials and beyond, supporting critical milestones toward commercial filing. This phase-appropriate, data-driven approach enables companies to meet regulatory requirements while optimizing the consistency, safety, and efficacy of their therapies.
References:
- U.S. Food and Drug Administration. (2023). Potency Assurance for Cellular and Gene Therapy Products: Draft Guidance for Industry. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/potency-assurance-cellular-and-gene-therapy-products
- European Medicines Agency. (2023). Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells (Revision 1). Retrieved from https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-and-clinical-aspects-medicinal-products-containing-genetically-modified-cells-revision-1_en.pdf
- U.S. Food and Drug Administration. (2011). Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products. Retrieved from https://www.fda.gov/files/vaccines,%20blood%20%26%20biologics/published/Final-Guidance-for-Industry--Potency-Tests-for-Cellular-and-Gene-Therapy-Products.pdf.
- Kiesgen S, Messinger JC, Chintala NK, Tano Z, Adusumilli PS. Comparative analysis of assays to measure CAR T-cell-mediated cytotoxicity. Nat Protoc. 2021 Mar;16(3):1331-1342. doi: 10.1038/s41596-020-00467-0. Epub 2021 Feb 15. PMID: 33589826; PMCID: PMC8064272.